Using goodtables to validate metadata from multiple sequencing runs
Introduction
I'm lucky to be working on my PhD as part of the Karstens' lab at Oregon Health and Science University. Our lab focuses on sequencing human samples that contain low amounts of bacterial DNA in an attempt to understand different types of bacterial communities and their effect on human health. We sequence hundreds of samples over many different sequencing experiments, yet we often need to combine these data and analyze them together. Depending on the experiment and questions we are asking, we collect and report different pieces of data and put them in a metadata file (csv).
These metadata files include relevant information about the sample, such as DNA concentration, origin of the sample, and an identifier specific to that sample. They often include other pieces of data specific to the experiment, such as if we're investigating the role of certain reagents and kits used and if they impact the bacteria found in each sample. Due to each metadata file having different types of information, it can be difficult to combine the files together for later analysis in R. This is where datapackages, schemas, and GoodTables validation comes in.
Goodtables is a data validator that checks both the structure and the contents of tabular data, like csvs. You can even enter in a schema to tell it what type of structure and contents to look for. This can be very helpful when comparing tabular data and making sure it matches, or in my case, can be combined.
Here, I will show you how I used a schema and GoodTables to make sure my metadata files could be combined, so I can use them for downstream microbial diversity analysis.
Using GoodTables for metadata
First, I created a datapackage using datapackagecreator and shown below, for my experiment labeled "MiSeq_Run_623", which stands for the 623rd sequencing run on an Illumina MiSeq machine. Next, I took only the "Fields" sections and created a schema to be used with the GoodTables browser tool.
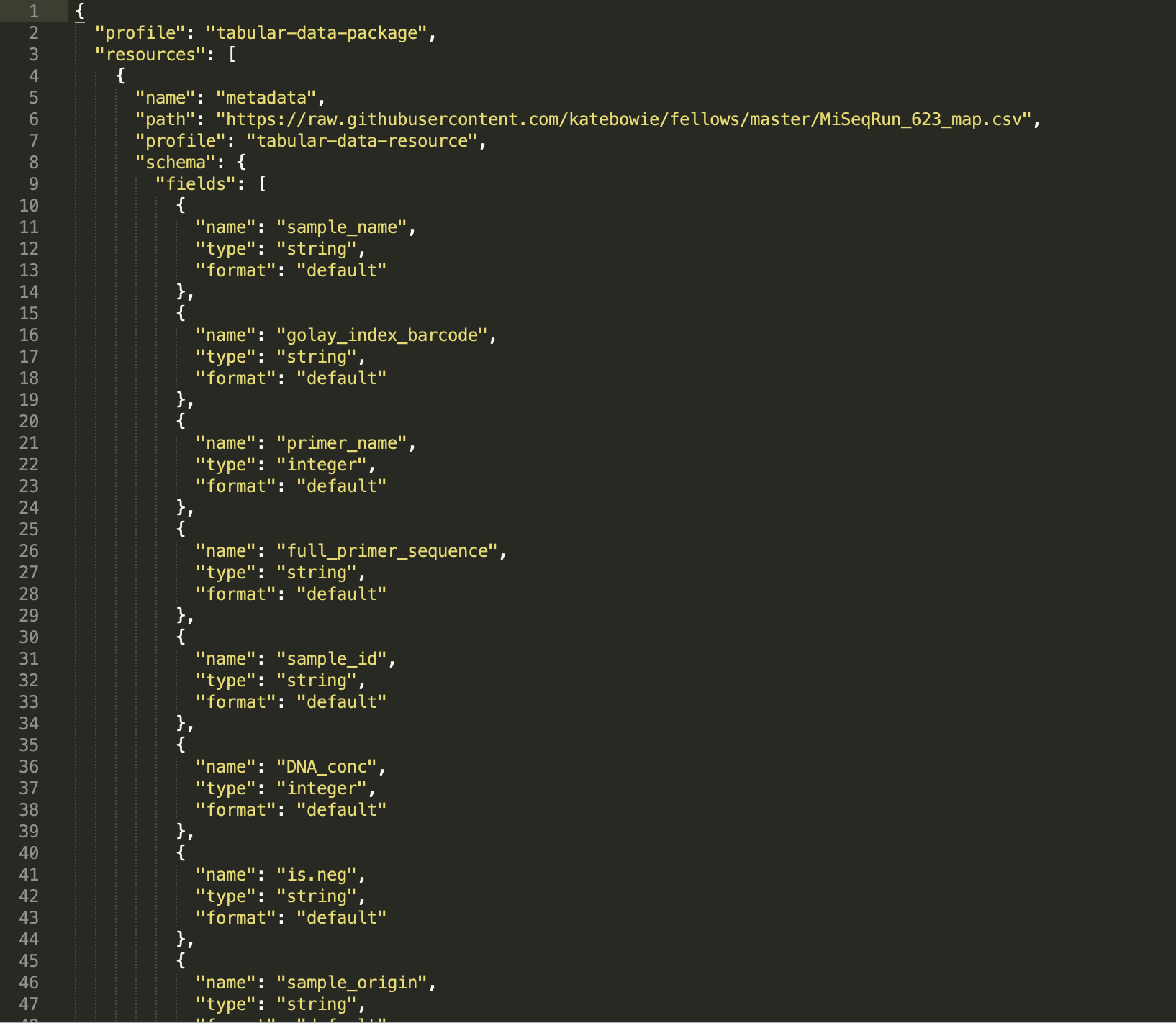
Now, I'm interested in joining this metadata from run 623, to metada from run 719. So I'm going to end up testing the metadata from MiSeq_Run_719 against the schema created for MiSeq_Run_623. First, I want to make sure that everything is working how it should be, and that I correctly created a schema. So I will be first using Goodtables to validate the metadata from the 623 run against the schema. (This should of course work, because the schema was created off of the 623 metadata file) And woo! It works!
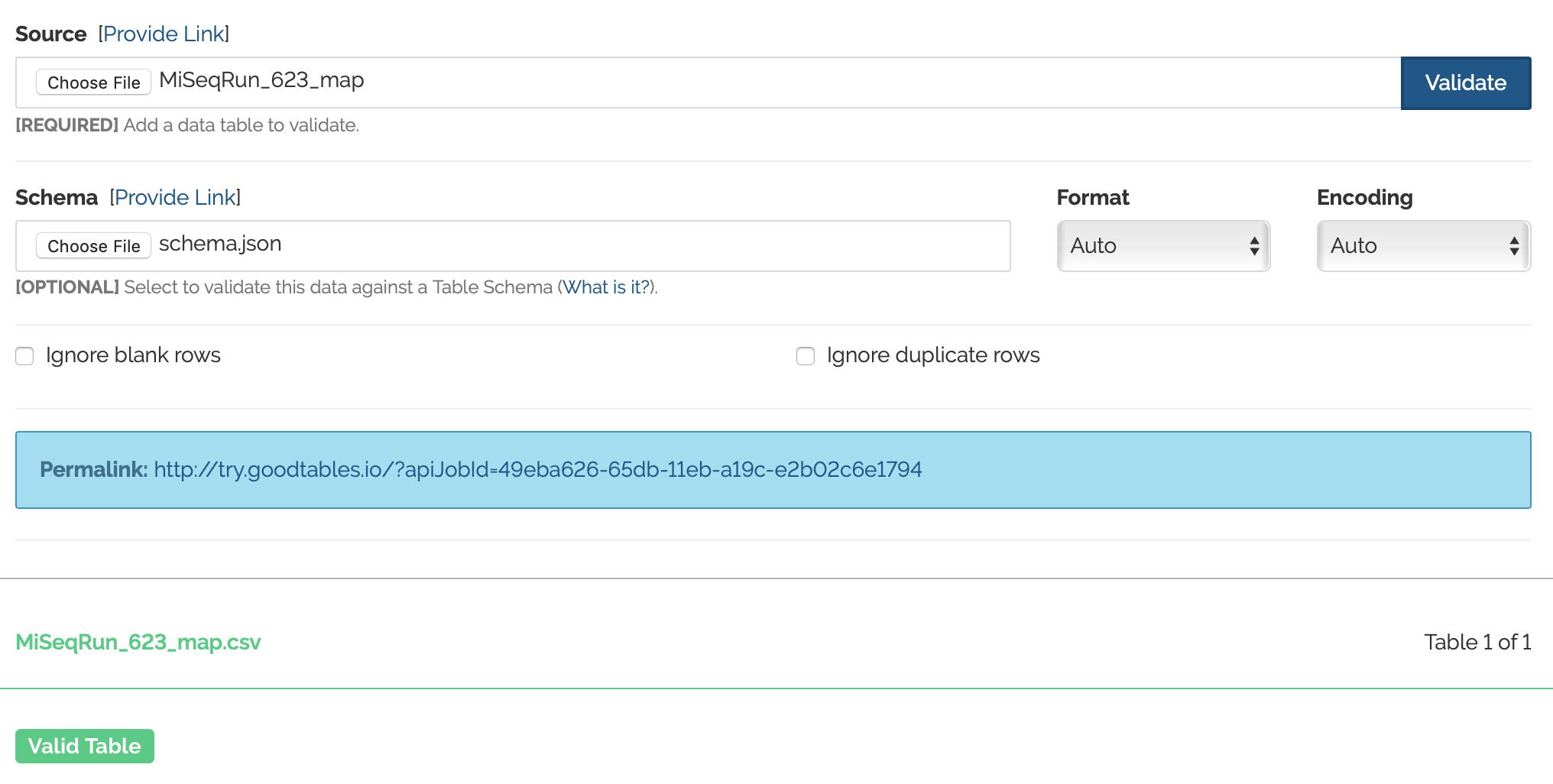
Next, I'm ready to validate the metadata from run 719 using the schema from run 623. As mentioned before, these are two different experiments and have very different sets of metadata reported, so I do not expect this to work.

The validation had many errors. Let's expand the errors to get an idea of what we're working with:

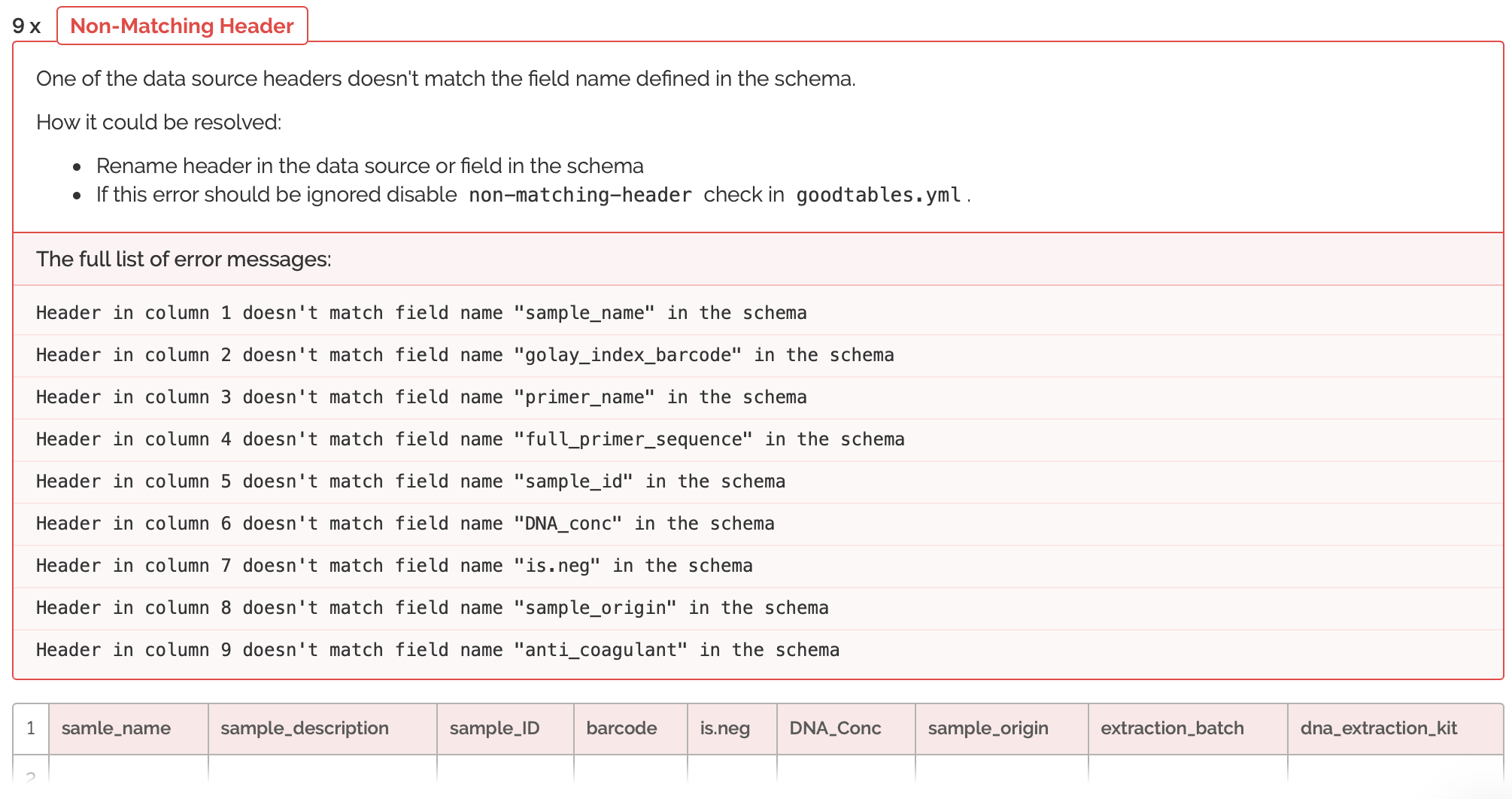
So here we're seeing that the columns are not only in the wrong order, but that many columns are missing. Also, there are many columns that do not match the schema provided by run 623. So, now I will have to do some switching around of columns. It's helpful, because the errors tell me exactly where my columns are not matching up and how I can fix this. After spending some time re-arranging, I'm going to try and validate the metadata from run 719 once more:
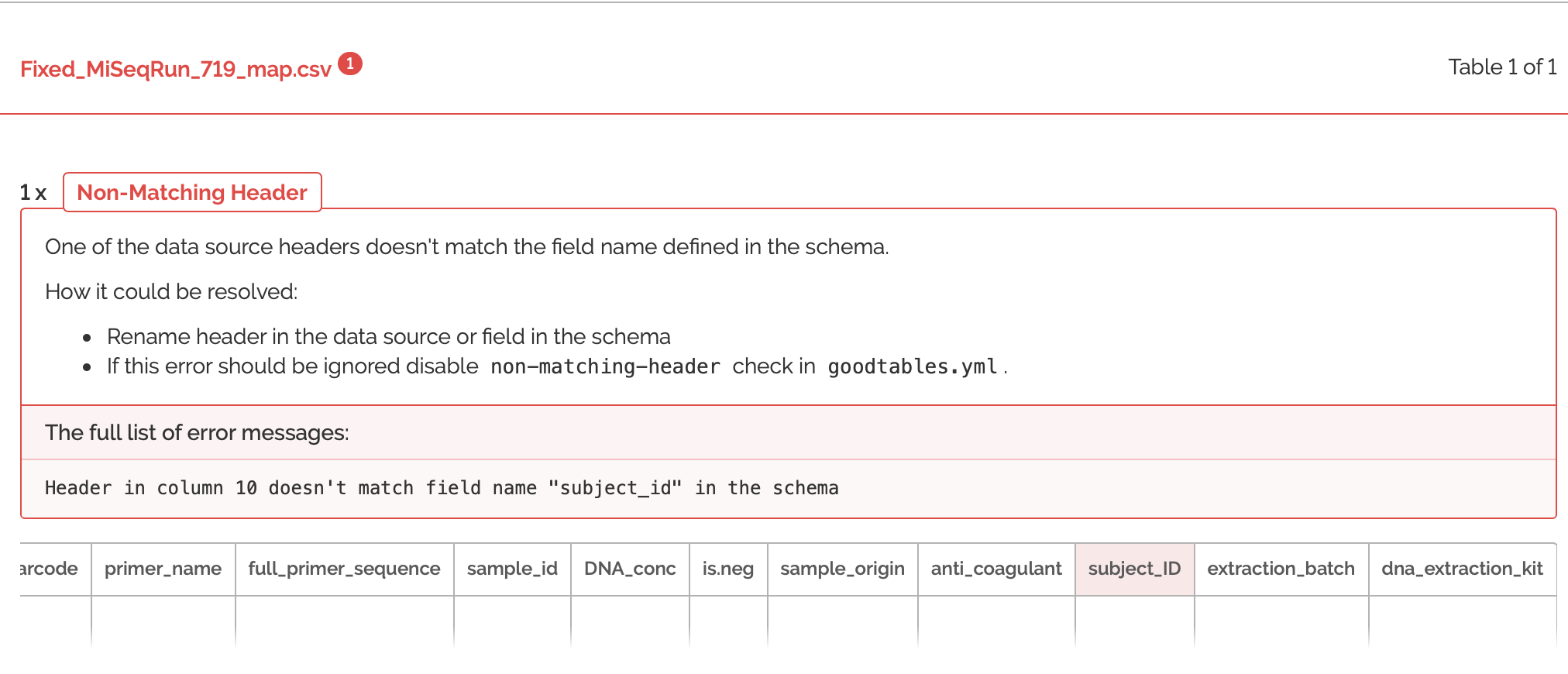
Ooh close, but not quite! It looks like I actually re-named that column "subjectid", instead of "subjectID". It's extremely helpful that GoodTables pointed this out, because if I tried to combine these metadatafiles in R with non-matching case as it is here, then it would create TWO separate columns for the metadata. Now, let's try to validate one more time with the fixed case of this header:

Success! Now I will be able to combine these metadata files together and it will make my data analysis pipeline a lot smoother. If interested in trying to validate these files yourself, please check them out on my github.